Hi all, I just started using GTDB and I am really impressed by this endeavor, wonderful work, and I’m excited to improve my working taxonomies!
I just ran the tool for the first time on some refseq complete genomes that I’m interested in, and it looks like the output tree has only the GTDB unique IDs or sometimes just the taxonomic rank plus taxa name (e.g. “g Leptolinea”). I’m wondering how I can associate these back to the original input IDs? I don’t see the GTDB ID column in the summary table either. Am I missing something?
Thanks a lot for clarification and continued work on this project. It’s a game changer.
-stephanie
Hi Stephanie,
If your input genomes has the name “MyGenome.fna” it will be called “MyGenome” in GTDB-Tk output tables and trees. This might be an issue with the program you are using to visualize the tree. I can recommend Dendroscope as being compatible with GTDB-Tk produced Newick files.
Cheers,
Donovan
Dear Donovan,
Thanks for your answer. I have looked at the tree file using a text editor and see the same issue that the nodes are named with only the GTDB IDs. This is the bac120.classify.tree file generated from the classify_wf command (no other tree files arrive in the final outputs so I presume this is the one expected). I’m working with iTol as my viewer, but I’ve also used figtree to double check, so I don’t think it’s an issue with the program. If the tree had given the input name I had for the genomes, there would be no issue. What I mean is that the tree gives an entirely new name (the gtdb unique id from the database) that I cannot associate back to my original IDs. Is this unexpected?
Thanks,
Stephanie
Hi Stephanie,
I’m not entirely following the issue here. GTDB-Tk does not change the names of input genomes or attempt to map them to the GTDB. All your query genomes passing QC should end up in the output with a name derived from the input filename (or the explicit name you gave them if using a batchfile). Perhaps you can send me an image and small dataset that illustrates the issue.
Cheers,
Donovan
Hi Stephanie,
I had the same problem, but figured it out.
The output from classify_wf makes a “decorated” tree, which includes “;” in the names of the samples. When the workflow makes a tree, the Newick format thinks “;” is part of the tree, not sample names, so iTOL and some other tree viewers don’t display the tree properly. You can use Dendroscope to view the classify_wf trees, OR:
Run the “infer” pipeline and view that tree on iTOL without decorating the tree with taxonomy.
This seems to be a bug, due to the taxonomy structure of GTDB. Donovan, is there any way to have an output option in the future that’s compatible with the iTOL viewer, because of its popularity? The GTDB-Tk pipeline is great and user-friendly, so having it compatible with iTOL and other tree viewers would be great.
Thanks,
Jason
Hi,
We have a lot of software written around the Newick format used by GTDB-Tk so it is a bit of a challenge to change this. Perhaps you can bring the issue up with iTOL. The trees produced by GTDB-Tk use quoted labels which should allow any characters to be used including a ‘;’. That said, the Newick format is poorly specified so I’m not surprised some tree viewers don’t handle quoted label.
Cheers,
Donovan
Dear all,
Sorry I stepped out of the convo the last weeks due to some travel and deadlines. I still don’t think my issue is clear or maybe I’m not clear on the expected files and output. But quickly - @rothmanj which file are you referring to as the decorated tree? Just to be clear.
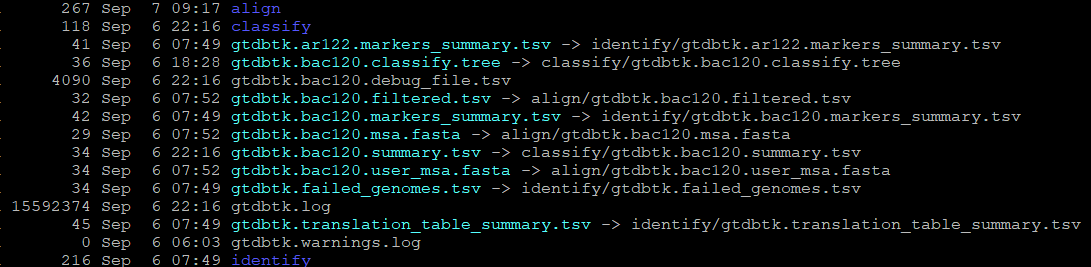
I’ll provide some screenshots and examples as best I can…
I installed in conda/bioconda and use the conda environment to run…
Here is my workflow call (forcing pplacer to use 1 cpu otherwise it errors and terminates):
WKDIR=/home/steph/COiB
4. Run the classify workflow
mkdir classify_out
conda activate gtdbtk
gtdbtk classify_wf \
--genome_dir $WKDIR/gtdb_analysis/refseq_fnas_461 \
--out_dir $WKDIR/gtdb_analysis/classify_out \
-x gz \
--cpus 8 \
--pplacer_cpus 1 \
--force \
--debug &
Here is the directory output. Based on the GTDB documentation, I understand that the “classify.tree” (or the “gtdbtk.bac120.classify.tree” as it is) is the tree of query sequences placed by pplacer.
I would expect this tree thus to have the names of my original queries, but it has only the GTDB IDs (GB_GCA_002347485.1 and etc…)
Maybe this is expected? Maybe this is just the bac120 reference genomes tree? But then I do not understand how that is helpful. I’m also still looking for where is the association key for my query genomes and the GTDB placement taxa name/ID (i.e. mygenome1 = GCA_123456789; mygenom2 = GCA_987654321… etc). I don’t see this provided in the summary.tsv (or “gtdbtk.bac120.summary.tsv” as it is).
I thought the identification and association to the new set of references provided by GTDB was a major motivation of this tool, so I’m assuming I completely misunderstand something and the output files here?
Thanks for your attention and time in helping.
Hi Stephanie,
Is this issue still outstanding or have you resolved your questions?
Briefly:
- yes, the
gtdbtk.bac120.classify.tree should contain your genome so long as it passed the GTDB-Tk QC criteria. A warning is produced indicating how many genomes fail QC.
- if your input genome is called
mygenome1 it will have this name in all GTDB-Tk output files
- the main output of the
classify workflow is the summary file which indicates the GTDB classification of your genome
The Newick trees (*.tree files) produced by GTDB-Tk follow a particular format. Some tree viewers are known to not fully parse this format. This includes iTOL. I recommend using Dendroscope to view the tree.
Cheers,
Donovan
Hi Donovan,
Still an outstanding issue. I do not seem to have the results as you describe, in that the gtdbtk.bac120.classify.tree does not contain my genomes’ names, only the gtdb IDs. This is regardless of tree viewer - I’m talking about when I manually look at the file itself in a text editor.
The query genomes I provide for the workflow are shown by name in the other output files, BUT, they do not associate with the gtdb IDs anywhere. There are only the gtdb taxonomic lineages provided, not the ID labels, and the ID labels are shown on the tree. Thus, I cannot associate the tree with the original sample files I provided. Does that make sense?
In short, while I want the GTDB classification of my genomes, I also want the gtdb ID that associates with that classification.
Thanks!
Hi Stephanie,
You query genomes do not get associated with a GTDB ID. Your genomes are placed into the GTDB reference tree and their taxonomic classification established based on this placement, their relative evolutionary divergence, and potentially ANI. For example, if a genome is placed within the Firmicutes phylum, but above the Bacilli class GTDB-Tk will classify it as p__Firmicutes and potentially c__Bacilli depending on the RED value of the genome. It never associated this genome with a GTDB ID as it is highly divergent from all reference genomes.
Do your genomes show up in the <prefix>.bac120.summary.tsv and/or <prefix>.ar122.summary.tsv files?
Details on GTDB-Tk can be found here:
Cheers,
Donovan
Hi,
Ah ok this is good to know. Yes my genomes show up in the summary.tsv files.
Regarding the tree - so if I understand you correctly, the tree shows the branches for the bac120 references along with my genomes, which are filed within the relevant nodes of this tree? That then explains why it is a mix of labels that are GTDB IDs and taxonomic labels, I think? That my own query genomes are sprinkled in amongst this reference tree?
Ah ha, yes. I see now. I’m sorry for the prolonged confusion (yes partly due to the difficulty in parsing the tree file and since I’m not using Dendroscope as you have recommended and no time to check it out at the moment). But thanks for sticking with me in clarifying this, I’m glad regardless of showing my extreme ignorance that now I think I have a very good understanding of how this works now. Keep up the GTDB work!
Cheers,
Stephanie
Hi @donovan.parks Thank you for the great tool firstly, I have a very similar question. I do understand from this thread that dendroscope is the best tool to use to parse adn view the gtdb tree output. I tried using dendroscope (just opened the gtdb tree file in dendroscope) and my tree has GB_GCA_… etc as tip labels. I tried iTOL and still the same.
I have some questions:
- Is there any specific way to using the tree file? Why does the phylogenetic tree not provide my sample names?
- How to add taxonomy and other metadata to the tree in this case?
Any help would be appreciated.
Thank you
DP
Hi Dhrati,
The gtdbtk.ar122.classify.tree and gtdbtk.ar122.classify.tree Newick tree files will contain any genomes you provided to GTDB-Tk that passed filtering. You can look at the GTDB-Tk log file to see how many genomes were placed into each domain tree and if any genomes failed the GTDB-Tk QC criteria. These Newick trees have GTDB taxon labels on internal nodes.
That said, these trees are provide to help users understand the classifications provided by GTDB-Tk. They are not de novo trees and do not follow best practice for phylogenetic inference so in general should not be used in publications.
Cheers,
Donovan
Hi @donovan.parks, thank you for your response. Sorry but I am confused, I am actually using the gtdbtk.bac120.classify.tree file to make the tree and my gtdbtk.warnings.log file formed after running gtdbtk is empty (so I assume everything passed the QC).
To be more precise, these are MAGs for three different groups. I would like to get MAG names on the tree instead of the GB_GCA…
What would you suggest in this case?
Thank you in advance,
Best
DP
Hi Dhrati,
I’m not sure what “MAG names” refers to. If your genome is named my_genome.fna it should appear in the gtdbtk.bac120.classify.tree as my_genome.fna (or perhaps my_genome, I can’t recall).
GB_GCA… are the IDs/names of the GTDB reference genomes. There is no mechanism to replace these with alternative names (species names?).
Hope this helps.
Cheers,
Donovan
Hi @donovan.parks Thank you for your response, by MAG I was referring to metagenome assembled genomes. Yes the name of my files is alright.
So basically my tree will have GTDB reference genomes as labels?
Thank you for clarifying this.
Best
DP
Hi.
The GTDB-Tk trees contain all GTDB reference genomes along with your genomes.
Cheers,
Donovan
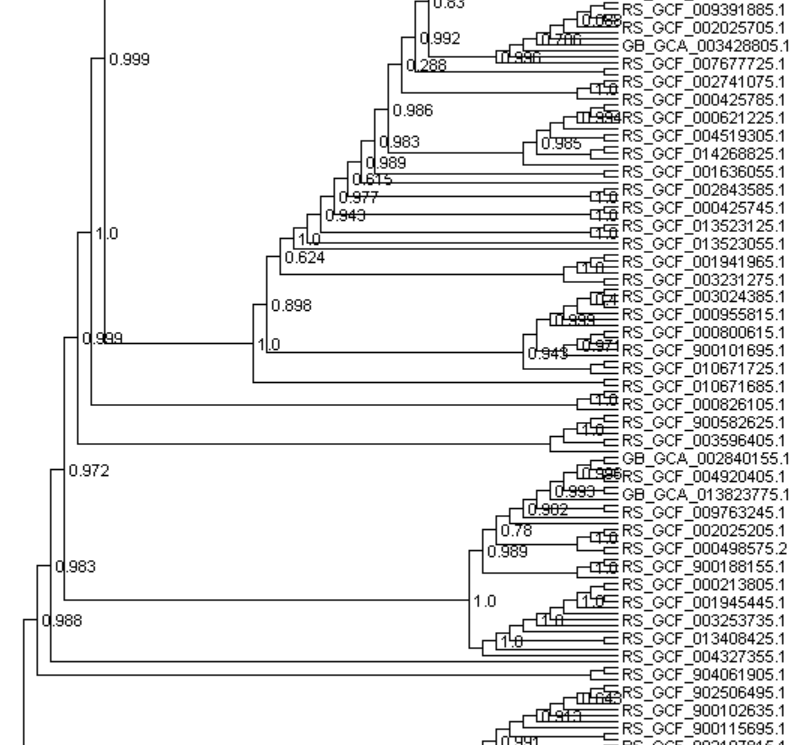
HI @donovan.parks thank you for your response, I am attaching a snip of the tree that I made from dendroscope for your reference. I generated this tree using the gtdbtk.bac120.classify.tree file. What I meant was how do I get my genome names or taxonomy instead of gtdb classifiers.
Apologies for the confusion
Thank you
Best
DP
Hi Dhrati,
Your genomes should be in this tree if you search for them. I’m not sure what you mean by getting your taxonomy into the tree. GTDB-Tk is based on the GTDB taxonomy and isn’t designed to handle other taxonomic sources. Perhaps if you mock up an image of what you were expecting to see I can comment further.
Cheers,
Donovan
Hi @donovan.parks you are right, some of my genome names are present in the tree. And by taxonomy I mean bacteria names for example instead of the gtdb ID. I found this other thread where someone provided a metadata file for gtdb IDs, can that be used to replace the IDs in the tree with bacteria names? NCBI Taxonomy Names in output · Issue #61 · Ecogenomics/GTDBTk · GitHub
Thank you very much for all your help
Best
DP