Hi,
Sorry for the dumb question, it is the first time I use GTDB-tk: I used gtdbtk classify_wf command, and got multiple treefiles in the output, and I could not figure out why, especially what is “gtdbtk.high.bac120.classify.tree”?
I got:
gtdbtk.bac120.classify.tree.11.tree
gtdbtk.bac120.classify.tree.6.tree
gtdbtk.high.bac120.classify.tree
gtdbtk.bac120.classify.tree.13.tree
gtdbtk.bac120.classify.tree.19.tree
Thanks in advance for the explanation.
Hello,
Starting with version 2.0.0, gtdbtk classify_wf pipeline uses the divide-and-conquer approach by default.
With this approach the placement of genomes is now divided in 2 steps:
- The first one is to place the user genomes onto a backbone tree representing one genome per family in GTDB (gtdbtk.high.bac120.classify.tree) .
- The genomes classified with a class level (order level for GTDB-Tk v2.0.0) on the backbone tree are then placed in a second tree (class-level subtrees) Those trees are made by a subsets of classes and are indexed for 1 to n ( gtdbtk.bac120.classify.tree..tree) .
- This second placement will finalise the taxonomy of the user genome down to species level ( if possible)
We are preparing a manuscript with the details.
Please note, from version 2.1.0, gtdbtk.high.bac120.classify.tree has been renamed to gtdbtk.backbone.bac120.classify.tree
Hello,
Thanks for the explanation. I was struggling with the same problem. i have one more doubt though.
I want to visualise the tree generated. in my tree.mapping.tsv file, i got 3 organisms that were mapped to classify.6.tree
when i am visualising i am getting following problems:
the tree generated has about 60 organisms while my file of classify.6.tree should have just 3.i don’t understand which one is mine.
my data is very new. recently sequenced.
thanks in advance.
Hi,
Can you attached the classify.6.tree and the mapping file in this thread?
Thanks
Hi,
i am attaching drive link. it has three files. one is summary of all the bacteria classified. another is the tree mapping file. and other one is classify.7.tree.
https://drive.google.com/drive/folders/1olbJI8l_dZRd_AqEaqMEltaqUuCdmYkR?usp=sharing
this is the tree that iam getting
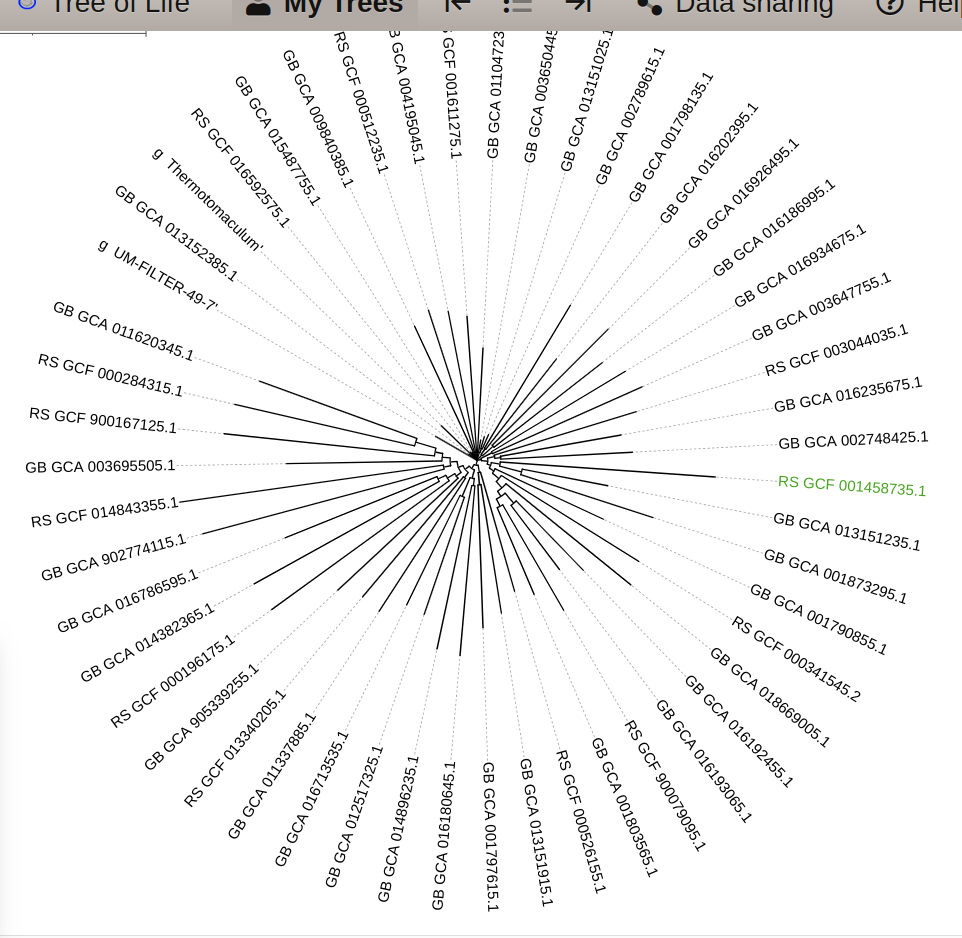
subset.7.tree (same as subset.6.tree) covers a subset of classes from the full GTDB reference tree. It consists of 9711 (10509 in subset.6.tree) reference genomes. so the output files will cover 9718 genomes for classify.7.tree ( 9711 + 7 user genomes based on the mapping file) and 10512 genomes for classify.6.tree ( 10509 + 3 user genomes based on the mapping file). Attached is a screenshot I have from classify7.tree where I highlight one of your genome (K_3_bin_13).
I would recommend using a tool like Dendroscope to open this tree files.
Hello I have a similar Question it’s my first time using GTDB-TK. i run the classify_wf on my MGAs. i have 82 of them i would like create a tree that only show my MAGs.
I would like to create something similar to what is done on this paper:
Fig. 2 | Scientific Data (nature.com)
here is my Classify_WF output:
i also have some doubt… this is the output tsv file from gtdbtk classify_wf.
| bin |
closest_genome_reference |
| bin.1.strict |
GCA_000435755.1 |
| bin.2.permissive |
GCF_025147485.1 |
| bin.4.strict |
GCF_025148445.1 |
| bin.5.strict |
GCF_002834235.1 |
| bin.6.permissive |
GCA_000432435.1 |
| bin.7.permissive |
GCF_003269275.1 |
| bin.8.strict |
GCA_003514385.1 |
In the tree, the IDs are something different, and I want to rename them based on the organism. How can I do this? The IDs shown in the trees are GB_GCA_027858855.1, GB_GCA_020721905.1, GB_GCA_001829495.1, GB_GCA_016934735.1, GB_GCA_029974015.1, GB_GCA_029562645.1, and GB_GCA_004297825.1."