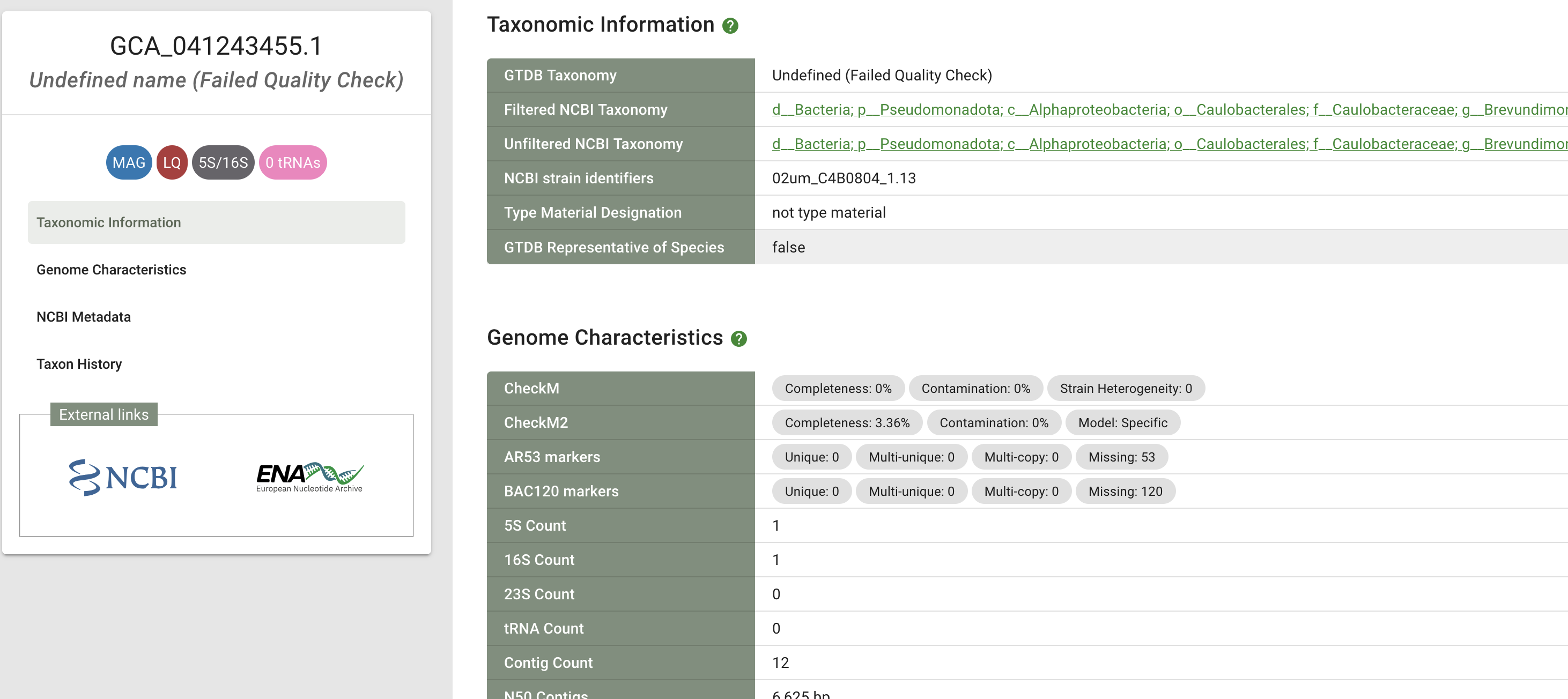
If I am reading the R226 release notes correctly, GTDB seems to apply filtering based on both CheckM1 and CheckM2, so what it accepts is just an intersection of what the two tools would accept. This goes a bit against this part of CheckM2 readme:
As a result of this machine learning framework, CheckM2 is also highly accurate on organisms with reduced genomes or unusual biology, such as the Nanoarchaeota or Patescibacteria.
Indeed there are genomes that score well on CheckM2 but not CheckM1 that are rejected by GTDB, including:
- GTDB - Loading... (cicada symbioant, Candidatus Hodgkinia)
- GTDB - Loading... (grasshopper symbioant, Vidania)
- GTDB - Loading... (also an insect symbioant, Nasuia)
I think the good CheckM2 completeness score and the contig count (1) provide enough confidence that the genome in question is indeed complete enough. Perhaps there should be a rule where GTDB considers genomes that score “particularly well” (perhaps > 70% complete, <5% contamination – I wanted to suggest a higher completeness bar but Vidania is still a little confusing to CheckM2 it seems) on CheckM2 to be acceptable regardless of what CheckM1 says.